Input/Output Formats
BDF Input Format
BDF supports three input formats: Easy Input, Advanced Input, and Mixed Input. Easy Input is user-friendly with a low barrier to entry, ideal for beginners. Advanced Input provides precise control over BDF modules. Mixed Input combines Easy Input with Advanced Input elements, offering convenience while enabling advanced functionality. Beginners can use Easy Input for most tasks, while users with quantum chemistry knowledge can leverage Advanced Input for deeper control.
Note
Input is case-insensitive except for filenames, shell commands, and environment variables. Module names, keywords, and values are case-insensitive.
BDF Easy Input
Example: Water molecule single-point energy calculation:
#!H2O.bdf
B3lyp/3-21G
Geometry # Atomic coordinates in Angstrom
O 0.00000 0.00000 0.36827
H 0.00000 -0.78398 -0.18468
H 0.00000 0.78398 -0.18468
End Geometry
Easy Input has three blocks:
First Block
Single line starting with #! followed by the script name (e.g., #!name.bdf). Can include descriptive text.
Second Block
From line 2 to the line before Geometry. Specifies calculation parameters (method, basis, functional, charge, multiplicity). Keywords are space-separated; values follow =. Multiple values use commas. Lines starting with # or content after # are comments.
Third Block
From Geometry to End Geometry. Defines molecular structure (see Molecular Structure Input Format).
Tip
Blank lines (except within
Geometry...End Geometry) are optional but improve readability.
BDF Advanced Input
Advanced Input uses a module-driven + parameter-control format:
$bdfmodule1
# Comment
Keyword1
value # inline comment
Keyword2
value
...
$end
%cp $BDFTASK.scforb $BDF_TMPDIR/$BDFTASK.inporb
$bdfmodule2
Keyword1
value
Keyword2
value
...
$end
- Description:
Modules execute sequentially. Each module starts with
$modulenameand ends with$end. Module names (e.g.,COMPASS,SCF) are defined in $BDFHOME/database/program.dat.Parameter Control: Keywords and values follow keyword + value format. Values start on the next line (single/multi-line).
Comments: Lines starting with
#or*, or content after#.Shell Commands: Lines starting with
%.Complex data blocks: Defined between
&database...&end(e.g., FLMO fragment definitions).
Example: Water molecule calculation:
#Example for BDF advanced input
$compass
Title
Water molecule, energy calculation
Geometry
O 0.00000 0.00000 0.36827
H 0.00000 -0.78398 -0.18468
H 0.00000 0.78398 -0.18468
End geometry
Basis # Basis set
3-21G
Group # C2v point group (auto-detected; used for D2h subgroups)
C(2v)
$end
$xuanyuan
$end
$scf
RHF # Restricted Hartree-Fock
$end
%cp $BDF_WORKDIR/$BDFTASK.scforb $BDF_TMPDIR/$BDFTASK.inporb
$scf
RKS # Restricted Kohn-Sham
DFT
B3lyp # Note: Differs from Gaussian's B3LYP
Guess
Readmo # Read orbitals as initial guess
$end
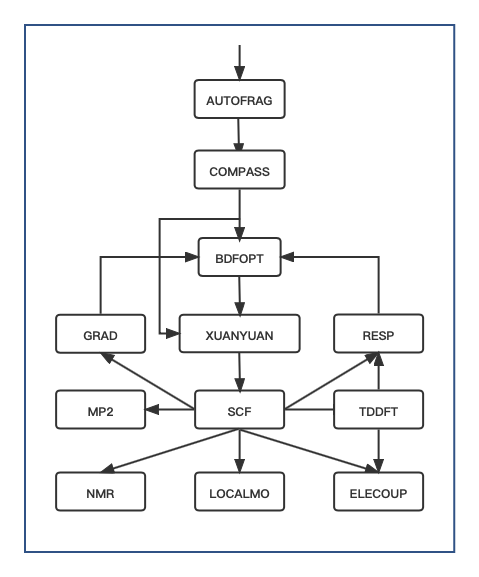
Fig. 1 BDF Module Execution Flow
Tip
Modules execute in order per the flow diagram. Most tasks use a subset (e.g., no
AUTOFRAGfor standard SCF).Complex tasks (e.g., geometry optimization) involve iterative module calls (e.g.,
XUANYUAN → SCF → RESP).Easy Input files are translated to Advanced Input in
$BDF_TMPDIR/.bdfinput.
Module |
Function |
|---|---|
AUTOFRAG |
Molecular fragmentation (iOI-SCF/FLMO) |
COMPASS |
Geometry, basis set, symmetry preprocessing |
XUANYUAN |
Atomic orbital integrals |
BDFOPT |
Geometry optimization |
SCF |
Hartree-Fock/Kohn-Sham SCF |
TDDFT |
Time-Dependent DFT |
RESP |
Gradients (HF/KS/TDDFT) |
GRAD |
Hartree-Fock gradients |
LOCALMO |
Molecular orbital localization |
NMR |
NMR shielding constants |
ELECOUP |
Electron coupling, localized excited states |
MP2 |
Møller-Plesset perturbation theory (MP2) |
Syntax |
Description |
|---|---|
$modulename…$end |
Module control block (modules in $BDFHOME/database/program.dat) |
# |
Line comment or inline comment (after #) |
Line comment (at start of line) |
|
% |
Shell command (executes after %) |
&database…&end |
Complex data block (e.g., FLMO fragments) |
BDF Mixed Input
Combines Easy Input convenience with Advanced Input precision:
#!name.bdf
Method/Functional/Basis Keyword=Option Keyword=Option1,Option2
Keyword=Option
Geometry
Molecular structure
End Geometry
$modulename1
... # Comment
$End
$modulename2
...
$End
- Structure:
Blocks 1-3: Easy Input format.
Block 4 (after
End Geometry): Advanced Input format (highest priority).
Example: Water cation:
#!H2O+.bdf
B3lyp/3-21G iroot=4
Geometry
O 0.00000 0.00000 0.36827
H 0.00000 -0.78398 -0.18468
H 0.00000 0.78398 -0.18468
End Geometry
$scf
Charge # Set charge to +1
1
molden # Output orbitals in Molden format
$end
Note: Advanced Input keywords override Easy Input settings (e.g., charge in $scf overrides command-line charge).
Molecular Structure Input Format
Molecular structure is defined between Geometry and End geometry. Formats: Cartesian coordinates, internal coordinates, or external XYZ file.
Warning
Default unit: Ångstrom (Å). Use unit=Bohr in Easy Input (command line) or Advanced Input (COMPASS module) for Bohr units.
Easy Input example (H₂ bond length: 1.50 Bohr):
#! bdftest.sh
HF/3-21G unit=Bohr
Geometry
H 0.00 0.00 0.00
H 0.00 0.00 1.50
End geometry
Advanced Input example:
$compass
Geometry
H 0.00 0.00 0.00
H 0.00 0.00 1.50
End geometry
Basis
3-21G
Unit
Bohr
$end
Cartesian Coordinates
Geometry # Default unit: Ångstrom
O 0.00000 0.00000 0.36937
H 0.00000 -0.78398 -0.18468
H 0.00000 0.78398 -0.18468
End geometry
Internal Coordinates (Z-Matrix)
Bond lengths (Å), angles/dihedrals (degrees):
Geometry
atom1
atom2 1 R12 # Bond length between atom2-atom1
atom3 1 R31 2 A312 # Bond R31, angle A312 (atoms 3-1-2)
atom4 3 R43 2 A432 1 D4321 # Bond R43, angle A432, dihedral D4321 (atoms 4-3-2-1)
...
End Geometry
Water example:
Geometry
O
H 1 0.9
H 1 0.9 2 109.0
End geometry
Variables (Easy Input only):
Geometry
O
H 1 R1
H 1 R1 2 A1
R1 = 0.9 # Variable definition
A1 = 109.0
End geometry
Warning
Separate variable definitions from coordinates with a blank line.
Potential Energy Scan (Easy Input only):
Example 1: H₂O bond scan (20 points, step 0.05 Å):
Geometry
O
H 1 R1
H 1 R1 2 109
R1 0.75 0.05 20 # Start, step, points
End geometry
Example 2: H₂O scan with SCF restart:
#! h2oscan.bdf
B3lyp/3-21G Scan Guess=readmo
Geometry
O
H 1 R1
H 1 R1 2 A1
A1 = 109.0
R1 0.75 0.05 20
End geometry
Read Coordinates from File
Geometry
file=filename.xyz # XYZ format file in current directory
End geometry
BDF Output Files
Additional temporary files may be generated for specific tasks.
Common Quantum Chemistry Units & Conversions
Quantum chemistry programs use atomic units (a.u.) internally. Outputs often convert to common units.
Energy: 1 a.u. = 1 Hartree
Mass: 1 a.u. = 1 mₑ (electron mass)
Length: 1 a.u. = 1 Bohr = 0.52917720859 Å
Charge: 1 a.u. = 1 e = 1.6022×10⁻¹⁹ C
Electron Density: 1 a.u. = 1 e/Bohr³
Dipole Moment: 1 a.u. = 1 e·Bohr = 2.5417462 Debye
Electrostatic Potential: 1 a.u. = 1 Hartree/e
Electric Field: 1 a.u. = 51421 V/Å
Energy Unit Conversions
1 unit = |
Hartree |
kJ·mol -1 |
kcal·mol -1 |
eV |
cm -1 |
Hartree |
1 |
2625.50 |
627.51 |
27.212 |
2.1947×10 5 |
kJ·mol -1 |
3.8088×10 -4 |
1 |
0.23901 |
1.0364×10 -2 |
83.593 |
kcal·mol -1 |
1.5936×10 -3 |
4.184 |
1 |
4.3363×10 -2 |
349.75 |
eV |
3.6749×10 -2 |
96.485 |
23.061 |
1 |
8065.5 |
cm -1 |
4.5563×10 -6 |
1.1963×10 -2 |
2.8591×10 -3 |
1.2398×10 -4 |
1 |
Length Unit Conversions
1 unit = |
Bohr |
Å |
nm |
Bohr |
1 |
0.52917720859 |
0.052917720859 |
Å |
1.88972613 |
1 |
0.1 |
nm |
0.188972613 |
10 |
1 |