Solvation Models
Solvation models are used to calculate interactions between solute and solvent, generally categorized into implicit solvent models (continuous medium models) and explicit solvent models. In BDF, for continuous solvent models, options include IEFPCM, SS(V)PE, CPCM, COSMO, ddCOSMO (domain-decomposition COSMO solvation model), and SMD. For explicit solvent models, the QM/MM method is employed, computed using the pDynamo2.0 program package.
Functionalities supported by BDF solvation models:
PCMs |
Ground state |
Excited state |
|||
|---|---|---|---|---|---|
Single-point |
Gradient |
Hessian |
Single-point |
Gradient |
|
COSMO |
√ |
√ |
√ |
√ |
√ |
CPCM |
√ |
√ |
√ |
√ |
√ |
SS(V)PE |
√ |
√ |
√ |
√ |
√ |
IEFPCM |
√ |
√ |
√ |
√ |
√ |
SMD |
√ |
√ |
√ |
√ |
√ |
Solvent Type Setting
Add the solvent keyword in the SCF module to enable solvation effect calculations. The solvent type (e.g., water) should be specified on the next line.
Example input for formaldehyde in aqueous solution:
$COMPASS
Title
ch2o Molecule test run
Basis
6-31g
Geometry
C 0.00000000 0.00000000 -0.54200000
O 0.00000000 0.00000000 0.67700000
H 0.00000000 0.93500000 -1.08200000
H 0.00000000 -0.93500000 -1.08200000
END geometry
nosymm
unit
ang
$END
$xuanyuan
$END
$SCF
rks
dft
b3lyp
solvent #Solvation calculation switch
water #Specify solvent
grid
medium
$END
Solvent types can be specified using names or aliases from BDF Supported Solvents List. For solvents not listed, input the dielectric constant:
solvent
user #User-specified
dielectric
78.3553 #Input dielectric constant
Solvent Model Setting
Continuous medium models treat the solvent as a polarizable continuous medium with a specific dielectric constant.
BDF currently supports ddCOSMO, COSMO, CPCM, IEFPCM, SS(V)PE, and SMD models. Keywords: ddcosmo, cosmo, cpcm, iefpcm, ssvpe, smd.
Input example:
solvent
water
solmodel
IEFPCM #Solvent model
For COSMO and CPCM, use cosmoFactorK to specify the dielectric screening factor \(f_\epsilon=\frac{\epsilon-1}{\epsilon+k}\). Default: k=0.5 for COSMO, k=0 for CPCM.
cosmoFactorK
0.5
For SMD, manually specify parameters:
refractiveIndex # Refractive index
1.43
HBondAcidity # Abraham hydrogen bond acidity
0.229
HBondBasicity # Abraham hydrogen bond basicity
0.265
SurfaceTensionAtInterface # Surface tension
61.24
CarbonAromaticity # Aromaticity
0.12
ElectronegativeHalogenicity # Halogenicity
0.24
Note
Using the SMD model disables calculation of non-electrostatic component of solvation free energy, replacing it with SMx series \(\Delta G_{CDS}\).
Cavity Customization
Cavity shape significantly impacts solvation energy. Common cavity types: vdW (van der Waals surface), SES (solvent-excluded surface), SAS (solvent-accessible surface).
BDF defaults to vdW cavity using 1.1× UFF radii. Customize cavity shape for COSMO/CPCM/IEFPCM/SS(V)PE/SMD using:
cavity # Cavity surface generation method
swig # swig | switching | ses | sphere (default: swig)
uatm # United atom topology method
false # false | true (default: false)
radiusType
UFF # UFF | Bondi (default: UFF)
vdWScale
1.1 # Default: 1.1 (1.1× RadiusType radius)
radii
1=1.4430 2=1.7500 # Set radius of atom 1 to 1.4430Å, atom 2 to 1.7500Å
# No spaces around "="; max 128 characters/line; multiple lines allowed
radii
H=1.4430 O=1.7500 # Set H radius to 1.4430Å, O to 1.7500Å (mix with above)
acidHRadius # Acidic H radius (Å)
1.2
Cavity Methods:
- switching: Smoothing function for vdW surface grid weights
- swig: Switching/Gaussian (additional Gaussian smoothing)
- sphere: Spherical cavity enclosing molecule
uatm merges H atoms into heavy atoms for cavity formation.
Control grid density with cavityNGrid or cavityPrecision:
cavityNGrid # Max tesserae per atom (adjusted to nearest Lebedev grid)
302 # Default: 302
# OR
cavityPrecision
medium # ultraCoarse | coarse | medium | fine | ultraFine (default: medium)
Ground State Solvation Energy Calculation
Typically requires only solvent and solmodel in SCF module.
Example for formaldehyde with SMD model:
$COMPASS
Title
ch2o Molecule test run
Basis
6-31g
Geometry
C 0.00000000 0.00000000 -0.54200000
O 0.00000000 0.00000000 0.67700000
H 0.00000000 0.93500000 -1.08200000
H 0.00000000 -0.93500000 -1.08200000
END geometry
$END
$xuanyuan
$END
$SCF
rks
dft
gb3lyp
solvent #Solvation switch
water #Solvent
solmodel #Solvation model
smd
$END
Note
Use cosmosave to export cavity volume/surface area, tesserae coordinates/charges/areas to .cosmo files. Convert to Gaussian format using $BDFHOME/sbin/conv2gaucosmo.py.
Non-Electrostatic Solvation Energy Calculation
Solvation free energy = Electrostatic (PCM) + Non-electrostatic (\(\Delta G_{cav}\) + \(\Delta G_{dis-rep}\)). Cavitation energy (work to form cavity) uses scaled particle theory (Pierotti-Claverie). Dispersion-repulsion uses pairwise potentials.
Non-electrostatic terms are disabled by default. Enable with:
nonels
dis rep cav # Dispersion | Repulsion | Cavitation
solventAtoms # Solvent atom counts (molecular formula)
H2O1 # Default: H2O1 ("1" required to avoid ambiguity)
solventRho # Solvent number density (molecules/ų)
0.03333
solventRadius # Solvent molecular radius (Å)
1.385
Note
For cav: Manual solventRho/solventRadius required for non-water solvents.
For dis/rep: Manual solventRho/solventAtoms required for non-water solvents.
Common Solvent Radii:
|----------------------|——-|-----------------|————-|----------|———|---------------------| | Radius (Å) | 1.385 | 2.900 | 2.815 | 1.855 | 2.180 | 2.685 |
Customize radii for dispersion-repulsion/cavitation:
solventAtomicSASRadii # SAS radii for dispersion-repulsion (solvent atoms)
H=1.20 O=1.50
radiiForCavEnergy # Radii for cavitation energy (solute)
H=1.4430 O=1.7500 # Same syntax as ``radii``
acidHRadiusForCavEnergy # Acidic H radius for cavitation (Å)
1.2
Introduction to Nonequilibrium Solvation Theory
Excited-state solvation requires nonequilibrium treatment due to rapid vertical absorption/emission processes. Solvent polarization has: - Fast (electronic) component - Slow (orientational) component
Traditional theories overestimate solvent reorganization energy. BDF implements new theory by Prof. Xiangyuan Li (Int. J. Quantum Chem. 2015, 115(11): 700-721) for state-specific calculations.
Excited State Solvation Effect Calculation
Implicit models handle excited states via: - Linear Response (LR) - State-Specific (SS)
### Vertical Absorption Calculation
Linear Response example:
$COMPASS
Title
ch2o Molecule test run
Basis
6-31g
Geometry
C 0.00000000 0.00000000 -0.54200000
O 0.00000000 0.00000000 0.67700000
H 0.00000000 0.93500000 -1.08200000
H 0.00000000 -0.9350000 -1.08200000
END geometry
nosymm
unit
ang
$END
$xuanyuan
$END
$SCF
rks
dft
b3lyp
grid
medium
solvent
user # User-specified
dielectric
78.3553 # Dielectric constant
opticalDielectric
1.7778 # Optical dielectric constant
solmodel
iefpcm
$END
$TDDFT
iroot
8
solneqlr # Enable nonequilibrium solvation (LR)
$END
Note
User-specified solvents require opticalDielectric (see BDF Supported Solvents List).
Perturbative State-Specific (ptSS) example:
$COMPASS
Title
SS-PCM of S-trans-acrolein Molecule
Basis
cc-PVDZ
Geometry
C 0.55794100 -0.45384200 -0.00001300
H 0.44564200 -1.53846100 -0.00002900
C -0.66970500 0.34745600 -0.00001300
H -0.50375600 1.44863100 -0.00005100
C 1.75266800 0.14414300 0.00001100
H 2.68187400 -0.42304000 0.00001600
H 1.83151500 1.23273300 0.00002700
O -1.78758800 -0.11830000 0.00001600
END geometry
$END
$xuanyuan
$END
$SCF
rks
dft
PBE0
solvent
water
solmodel
iefpcm
$END
$TDDFT
iroot
5
istore
1
$END
$resp
nfiles
1
method
2
iroot
1 2 3
geom
norder
0
solneqss # State-specific nonequilibrium
$end
Output snippet:
-Energy correction based on constrant equilibrium theory with relaxed density
*State 1 -> 0
Corrected vertical absorption energy = 3.6935 eV
Nonequilibrium solvation free energy = -0.0700 eV
Equilibrium solvation free energy = -0.1744 eV
Among them, Corrected vertical absorption energy refers to the excitation energy correction calculated by using the new theory of non-equilibrium solvation developed by Prof. Xiangyuan Li’s group.
In the above example, the vertical absorption energy is \(3.69eV\).
BDF currently also supports the calculation of corrected linear response (cLR), and the following is an input file for calculating the non-equilibrium solvation effect of acrolein molecules in the excited state using cLR:
$COMPASS
Title
cLR-PCM of S-trans-acrolein Molecule
Basis
cc-PVDZ
Geometry
C 0.55794100 -0.45384200 -0.00001300
H 0.44564200 -1.53846100 -0.00002900
C -0.66970500 0.34745600 -0.00001300
H -0.50375600 1.44863100 -0.00005100
C 1.75266800 0.14414300 0.00001100
H 2.68187400 -0.42304000 0.00001600
H 1.83151500 1.23273300 0.00002700
O -1.78758800 -0.11830000 0.00001600
END geometry
$END
$xuanyuan
$END
$SCF
rks
dft
PBE0
solvent
water
solmodel
iefpcm
$END
$TDDFT
iroot
5
istore
1
$END
$TDDFT
iroot
5
istore
1
solneqlr
$END
$resp
nfiles
1
method
2
iroot
1
geom
norder
0
solneqlr
solneqss
$end
Locate the output from the first TDDFT section and the cLR output from the resp module:
No. 1 w= 3.7475 eV -191.566549 a.u. f= 0.0001 D<Pab>= 0.0000 Ova= 0.4683
CV(0): A( 15 )-> A( 16 ) c_i: 0.9871 Per: 97.4% IPA: 5.808 eV Oai: 0.4688
CV(0): A( 15 )-> A( 17 ) c_i: 0.1496 Per: 2.2% IPA: 9.144 eV Oai: 0.4392
...
Excitation energy correction(cLR) = -0.0377 eV
The cLR excitation energy is calculated as: \(3.7475 - 0.0377 = 3.7098\text{eV}\).
### Excited State Geometry Optimization
During geometry optimization, solvent has sufficient time to respond, so equilibrium solvation should be considered.
Use the soleqlr keyword in both tddft and resp modules to enable equilibrium solvation effects. Other input/output details are similar to the TDDFT geometry optimization section and won’t be repeated here.
Phenol molecule example with solvation effects:
$COMPASS
Geometry
C -1.15617700 -1.20786100 0.00501300
C -1.85718200 0.00000000 0.01667700
C -1.15617700 1.20786100 0.00501300
C 0.23962700 1.21165300 -0.01258600
C 0.93461900 0.00000000 -0.01633400
C 0.23962700 -1.21165300 -0.01258600
H -1.69626800 -2.15127300 0.00745900
H -2.94368500 0.00000000 0.02907200
H -1.69626800 2.15127300 0.00745900
H 0.80143900 2.14104700 -0.03186000
H 0.80143900 -2.14104700 -0.03186000
O 2.32295900 0.00000000 -0.08796400
H 2.68364400 0.00000000 0.81225800
End geometry
basis
6-31G
$END
$bdfopt
solver
1
$end
$XUANYUAN
$END
$SCF
DFT
gb3lyp
rks
solModel
iefpcm
solvent
water
$END
$TDDFT
iroot
5
istore
1
soleqlr
$END
$resp
geom
soleqlr
method
2
nfiles
1
iroot
1
$end
Vertical Emission Calculation
In the equilibrium geometry of the excited state, the equilibrium solvation effect of ptSS or cLR is calculated, and the corresponding solvent slow polarization charge is saved. The keyword ‘’emit’’ was added to the SCF module to calculate the non-equilibrium ground state energy. Taking the acrolein molecule as an example, ptSS is used to calculate the excited state, and the corresponding input file is as follows:
$COMPASS
Geometry
C -1.810472 0.158959 0.000002
H -1.949516 1.241815 0.000018
H -2.698562 -0.472615 -0.000042
C -0.549925 -0.413873 0.000029
H -0.443723 -1.502963 -0.000000
C 0.644085 0.314498 0.000060
H 0.618815 1.429158 -0.000047
O 1.862127 -0.113145 -0.000086
End geometry
basis
cc-PVDZ
$END
$XUANYUAN
$END
$SCF
DFT
PBE0
rks
solModel
iefpcm
solvent
water
$END
$TDDFT
iroot
5
istore
1
#soleqlr
$END
$resp
nfiles
1
method
2
iroot
1
geom
norder
0
#soleqlr
soleqss
$end
$SCF
DFT
PBE0
rks
solModel
iefpcm
solvent
water
emit
$END
Care needs to be taken to specify ‘’soleqss’’ to calculate the equilibrium solvation effect. The output in the file is:
-Energy correction based on constrant equilibrium theory
*State 1 -> 0
Corrected vertical emission energy = 2.8118 eV
Nonequilibrium solvation free energy = -0.0964 eV
Equilibrium solvation free energy = -0.1145 eV
The “Corrected vertical emission energy” represents the emission energy correction using Prof. Li’s theory. In this example, vertical emission energy is \(2.81eV\).
When using the cLR calculation, you need to find the output of the first TDDFT in the file, and the cLR output in the resp module, and add it to the difference between the E_tot two scfs to get the final vertical emission energy.
A combination of explicit and implicit solvents was used to calculate the aroused solvation effect
The excited solvation effect can be calculated using a combination of explicit and implicit solvents. In the case of aqueous solutions, it is possible to diffuse to the HOMO and LUMO orbitals of the solute molecules The first hydrate layer, so the water molecules of the first hydrate can be included in the TDDFT calculation area when performing the excited state calculation, while the rest is treated with implicit solvents.
Take sinapic acid, for example. To determine the first hydrate layer of solute molecules, the Amber procedure can be used to perform molecular dynamics simulations by placing the sinapic acid molecules in small water boxes. After the system is equilibrated, the distribution of water molecules around the solute molecules can be analyzed to determine the first hydration layer. Of course, it is also possible to select a multi-frame structure for calculation and then average it.
Hydrate molecule selection can be done using the VMD program. Assuming the input is a pdb file, the first hydrate molecule can be selected in the command line and saved as a pdb file. The command is as follows:
atomselect top "same resid as (within 3.5 of not water)" # Select the first hydrate layer
atomselect0 writepdb sa.pdb #The solute molecule and the first hydrate layer are stored in a PDB file
In the example above, all water molecules within 3.5 angstroms of the solute molecule are selected, and as long as one of the three atoms of the water molecule is within the truncated range, the entire molecule is selected. The selection result is shown in the figure:
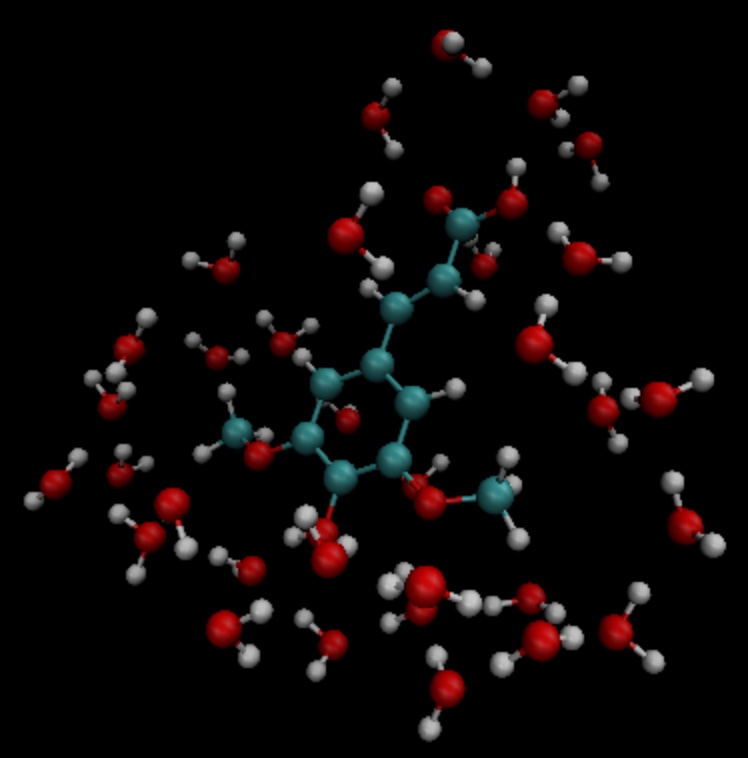
According to the coordinate information in the sa.pdb file, the TDDFT is calculated, and the input file is as follows:
$COMPASS
Title
SA Molecule test run
Basis
6-31g
Geometry
C 14.983 14.539 6.274
C 14.515 14.183 7.629
C 13.251 14.233 8.118
C 12.774 13.868 9.480
C 11.429 14.087 9.838
C 10.961 13.725 11.118
O 9.666 13.973 11.525
C 8.553 14.050 10.621
C 11.836 13.125 12.041
O 11.364 12.722 13.262
C 13.184 12.919 11.700
O 14.021 12.342 12.636
C 15.284 11.744 12.293
C 13.648 13.297 10.427
O 14.270 14.853 5.341
O 16.307 14.468 6.130
H 15.310 13.847 8.286
H 12.474 14.613 7.454
H 10.754 14.550 9.127
H 7.627 14.202 11.188
H 8.673 14.888 9.924
H 8.457 13.118 10.054
H 10.366 12.712 13.206
H 15.725 11.272 13.177
H 15.144 10.973 11.525
H 15.985 12.500 11.922
H 14.687 13.129 10.174
H 16.438 14.756 5.181
O 18.736 9.803 12.472
H 18.779 10.597 11.888
H 19.417 10.074 13.139
O 18.022 14.021 8.274
H 17.547 14.250 7.452
H 18.614 13.310 7.941
O 8.888 16.439 7.042
H 9.682 16.973 6.797
H 8.217 17.162 7.048
O 4.019 14.176 11.140
H 4.032 13.572 10.360
H 4.752 14.783 10.885
O 16.970 8.986 14.331
H 17.578 9.273 13.606
H 17.497 8.225 14.676
O 8.133 17.541 10.454
H 8.419 17.716 11.386
H 8.936 17.880 9.990
O 8.639 12.198 13.660
H 7.777 11.857 13.323
H 8.413 13.155 13.731
O 13.766 11.972 4.742
H 13.858 12.934 4.618
H 13.712 11.679 3.799
O 10.264 16.103 14.305
H 9.444 15.605 14.054
H 10.527 15.554 15.084
O 13.269 16.802 3.701
H 13.513 16.077 4.325
H 14.141 17.264 3.657
O 13.286 14.138 14.908
H 13.185 14.974 14.393
H 13.003 13.492 14.228
O 16.694 11.449 15.608
H 15.780 11.262 15.969
H 16.838 10.579 15.161
O 7.858 14.828 14.050
H 7.208 15.473 13.691
H 7.322 14.462 14.795
O 15.961 17.544 3.706
H 16.342 16.631 3.627
H 16.502 17.866 4.462
O 10.940 14.245 16.302
H 10.828 13.277 16.477
H 11.870 14.226 15.967
O 12.686 10.250 14.079
H 11.731 10.151 14.318
H 12.629 11.070 13.541
O 9.429 11.239 8.483
H 8.927 10.817 7.750
H 9.237 12.182 8.295
O 17.151 15.141 3.699
H 17.124 14.305 3.168
H 18.133 15.245 3.766
O 17.065 10.633 9.634
H 16.918 10.557 8.674
H 17.024 9.698 9.909
O 17.536 14.457 10.874
H 18.014 13.627 11.089
H 17.683 14.460 9.890
O 5.836 16.609 13.299
H 4.877 16.500 13.549
H 5.760 16.376 12.342
O 19.014 12.008 10.822
H 18.249 11.634 10.308
H 19.749 11.655 10.256
O 15.861 14.137 15.750
H 14.900 13.990 15.574
H 16.185 13.214 15.645
O 11.084 9.639 10.009
H 11.641 9.480 9.213
H 10.452 10.296 9.627
O 14.234 10.787 16.235
H 13.668 10.623 15.444
H 13.663 10.376 16.925
O 14.488 8.506 13.105
H 13.870 9.136 13.550
H 15.301 8.683 13.628
O 14.899 17.658 9.746
H 15.674 18.005 9.236
H 15.210 16.754 9.926
O 8.725 13.791 7.422
H 9.237 13.488 6.631
H 8.845 14.770 7.309
O 10.084 10.156 14.803
H 9.498 10.821 14.366
H 10.215 10.613 15.669
O 5.806 16.161 10.582
H 5.389 16.831 9.993
H 6.747 16.470 10.509
O 6.028 13.931 7.206
H 5.971 14.900 7.257
H 6.999 13.804 7.336
O 17.072 12.787 2.438
H 16.281 12.594 1.885
H 17.062 11.978 3.013
END geometry
nosymm
mpec+cosx
$END
$xuanyuan
$end
$SCF
rks
dft
b3lyp
solvent
water
grid
medium
$END
# input for tddft
$tddft
iroot # Calculate 1 root for each irrep. By default, 10 roots are calculated
1 # for each irrep
memjkop # maxium memeory for Coulomb and Exchange operator. 1024 MW (Mega Words)
1024
$end
A list of solvent types supported in BDF
Name |
Short Name |
\({\epsilon}\) |
\({\epsilon_{opt}}\) |
|---|---|---|---|
water |
H2O |
78.3553 |
1.7764 |
acetic acid |
ACETACID |
6.2528 |
1.8824 |
acetone |
ACETONE |
20.4930 |
1.8463 |
acetonitrile |
ACETNTRL |
35.6880 |
1.8069 |
acetophenone |
ACETPHEN |
17.4400 |
2.3630 |
aniline |
ANILINE |
6.8882 |
2.5163 |
anisole |
ANISOLE |
4.2247 |
2.3025 |
benzaldehyde |
BENZALDH |
18.2200 |
2.3910 |
benzene |
BENZENE |
2.2706 |
2.2533 |
benzonitrile |
BENZNTRL |
25.5920 |
2.3375 |
benzyl chloride |
BENZYLCL |
6.7175 |
2.3688 |
1-bromo-2-methylpropane |
BRISOBUT |
7.7792 |
2.0587 |
bromobenzene |
BRBENZEN |
5.3954 |
2.4327 |
bromoethane |
BRETHANE |
9.0100 |
2.0275 |
bromoform |
BROMFORM |
4.2488 |
2.5616 |
1-bromooctane |
BROCTANE |
5.0244 |
2.1095 |
1-bromopentane |
BRPENTAN |
6.2690 |
2.0872 |
2-bromopropane |
BRPROPA2 |
9.3610 |
2.0309 |
1-bromopropane |
BRPROPAN |
8.0496 |
2.0572 |
butanal |
BUTANAL |
13.4500 |
1.9163 |
butanoic acid |
BUTACID |
2.9931 |
1.9544 |
1-butanol |
BUTANOL |
17.3320 |
1.9580 |
2-butanol |
BUTANOL2 |
15.9440 |
1.9538 |
butanone |
BUTANONE |
18.2460 |
1.9011 |
butanonitrile |
BUTANTRL |
24.2910 |
1.9160 |
butyl acetate |
BUTILE |
4.9941 |
1.9435 |
butylamine |
NBA |
4.6178 |
1.9687 |
n-butylbenzene |
NBUTBENZ |
2.3600 |
2.2195 |
sec-butylbenzene |
SBUTBENZ |
2.3446 |
2.2186 |
tert-butylbenzene |
TBUTBENZ |
2.3447 |
2.2282 |
carbon disulfide |
CS2 |
2.6105 |
2.6631 |
carbon tetrachloride |
CARBNTET |
2.2280 |
2.1319 |
chlorobenzene |
CLBENZEN |
5.6968 |
2.3229 |
sec-butyl chloride |
SECBUTCL |
8.3930 |
1.9519 |
chloroform |
CHCL3 |
4.7113 |
2.0906 |
1-chlorohexane |
CLHEXANE |
5.9491 |
2.0161 |
1-chloropentane |
CLPENTAN |
6.5022 |
1.9957 |
1-chloropropane |
CLPROPAN |
8.3548 |
1.9263 |
o-chlorotoluene |
OCLTOLUE |
4.6331 |
2.3311 |
m-cresol |
M-CRESOL |
12.4400 |
2.3833 |
o-cresol |
O-CRESOL |
6.7600 |
2.3596 |
cyclohexane |
CYCHEXAN |
2.0165 |
2.0352 |
cyclohexanone |
CYCHEXON |
15.6190 |
2.1045 |
cyclopentane |
CYCPENTN |
1.9608 |
1.9782 |
cyclopentanol |
CYCPNTOL |
16.9890 |
2.1112 |
cyclopentanone |
CYCPNTON |
13.5800 |
2.0638 |
cis-decalin |
DECLNCIS |
2.2139 |
2.1934 |
trans-decalin |
DECLNTRA |
2.1781 |
2.1594 |
decalin (cis/trans mixture) |
DECLNMIX |
2.1960 |
2.1765 |
n-decane |
DECANE |
1.9846 |
1.9887 |
1-decanol |
DECANOL |
7.5305 |
2.0655 |
1,2-dibromoethane |
EDB12 |
4.9313 |
2.3676 |
dibromomethane |
DIBRMETN |
7.2273 |
2.3778 |
dibutyl ether |
BUTYLETH |
3.0473 |
1.9578 |
o-dichlorobenzene |
ODICLBNZ |
9.9949 |
2.4072 |
1,2-dichloroethane |
EDC12 |
10.1250 |
2.0874 |
cis-dichloroethylene |
C12DCE |
9.2000 |
2.0996 |
trans-dichloroethylene |
T12DCE |
2.1400 |
2.0892 |
dichloromethane |
DCM |
8.9300 |
2.0283 |
diethyl ether |
ETHER |
4.2400 |
1.8295 |
diethyl sulfide |
ET2S |
5.7230 |
2.0822 |
diethylamine |
DIETAMIN |
3.5766 |
1.9221 |
diiodomethane |
MI |
5.3200 |
3.0363 |
diisopropyl ether |
DIPE |
3.3800 |
1.8712 |
dimethyl disulfide |
DMDS |
9.6000 |
2.3375 |
dimethyl sulfoxide |
DMSO |
46.8260 |
2.0079 |
n,n-dimethylacetamide |
DMA |
37.7810 |
2.0678 |
cis-1,2-dimethylcyclohexane |
CISDMCHX |
2.0600 |
2.0621 |
n,n-dimethylformamide |
DMF |
37.2190 |
2.0463 |
2,4-dimethylpentane |
DMEPEN24 |
1.8939 |
1.9085 |
2,4-dimethylpyridine |
DMEPYR24 |
9.4176 |
2.2530 |
2,6-dimethylpyridine |
DMEPYR26 |
7.1735 |
2.2359 |
1,4-dioxane |
DIOXANE |
2.2099 |
2.0232 |
diphenyl ether |
PHOPH |
3.7300 |
2.4923 |
dipropylamine |
DPROAMIN |
2.9112 |
1.9740 |
n-dodecane |
DODECAN |
2.0060 |
2.0209 |
1,2-ethanediol |
MEG |
40.2450 |
2.0501 |
ethanethiol |
ETSH |
6.6670 |
2.0478 |
ethanol |
ETHANOL |
24.8520 |
1.8526 |
ethyl acetate |
ETOAC |
5.9867 |
1.8832 |
ethyl formate |
ETOME |
8.3310 |
1.8493 |
ethylbenzene |
EB |
2.4339 |
2.2377 |
ethylphenyl ether |
PHENETOL |
4.1797 |
2.2729 |
fluorobenzene |
C6H5F |
5.4200 |
2.1562 |
1-fluorooctane |
FOCTANE |
3.8900 |
1.9418 |
formamide |
FORMAMID |
108.9400 |
2.0944 |
formic acid |
FORMACID |
51.1000 |
1.8807 |
n-heptane |
HEPTANE |
1.9113 |
1.9260 |
1-heptanol |
HEPTANOL |
11.3210 |
2.0303 |
2-heptanone |
HEPTNON2 |
11.6580 |
1.9847 |
4-heptanone |
HEPTNON4 |
12.2570 |
1.9794 |
n-hexadecane |
HEXADECN |
2.0402 |
2.0578 |
n-hexane |
HEXANE |
1.8819 |
1.8904 |
hexanoic acid |
HEXNACID |
2.6000 |
2.0059 |
1-hexanol |
HEXANOL |
12.5100 |
2.0102 |
2-hexanone |
HEXANON2 |
14.1360 |
1.9620 |
1-hexene |
HEXENE |
2.0717 |
1.9146 |
1-hexyne |
HEXYNE |
2.6150 |
1.9569 |
iodobenzene |
C6H5I |
4.5470 |
2.6244 |
1-iodobutane |
IOBUTANE |
6.1730 |
2.2503 |
iodoethane |
C2H5I |
7.6177 |
2.2901 |
1-iodohexadecane |
IOHEXDEC |
3.5338 |
2.1922 |
iodomethane |
CH3I |
6.8650 |
2.3654 |
1-iodopentane |
IOPENTAN |
5.6973 |
2.2377 |
1-iodopropane |
IOPROPAN |
6.9626 |
2.2674 |
isopropylbenzene |
CUMENE |
2.3712 |
2.2246 |
p-isopropyltoluene |
P-CYMENE |
2.2322 |
2.2228 |
mesitylene |
MESITYLN |
2.2650 |
2.2482 |
methanol |
METHANOL |
32.6130 |
1.7657 |
2-methoxyethanol |
EGME |
17.2000 |
1.9667 |
methyl acetate |
MEACETAT |
6.8615 |
1.8534 |
methyl benzoate |
MEBNZATE |
6.7367 |
2.2995 |
methyl butanoate |
MEBUTATE |
5.5607 |
1.9260 |
methyl formate |
MEFORMAT |
8.8377 |
1.8045 |
4-methyl-2-pentanone |
MIBK |
12.8870 |
1.9494 |
methyl propanoate |
MEPROPYL |
6.0777 |
1.8975 |
2-methyl-1-propanol |
ISOBUTOL |
16.7770 |
1.9474 |
2-methyl-2-propanol |
TERBUTOL |
12.4700 |
1.9260 |
n-methylaniline |
NMEANILN |
5.9600 |
2.4599 |
methylcyclohexane |
MECYCHEX |
2.0240 |
2.0252 |
n-methylformamide (E/Z mixture) |
NMFMIXTR |
181.5600 |
2.0503 |
2-methylpentane |
ISOHEXAN |
1.8900 |
1.8810 |
2-methylpyridine |
MEPYRID2 |
9.9533 |
2.2371 |
3-methylpyridine |
MEPYRID3 |
11.6450 |
2.2620 |
4-methylpyridine |
MEPYRID4 |
11.9570 |
2.2611 |
nitrobenzene |
C6H5NO2 |
34.8090 |
2.4218 |
nitroethane |
C2H5NO2 |
28.2900 |
1.9368 |
nitromethane |
CH3NO2 |
36.5620 |
1.9091 |
1-nitropropane |
NTRPROP1 |
23.7300 |
1.9650 |
2-nitropropane |
NTRPROP2 |
25.6540 |
1.9444 |
o-nitrotoluene |
ONTRTOLU |
25.6690 |
2.3870 |
n-nonane |
NONANE |
1.9605 |
1.9751 |
1-nonanol |
NONANOL |
8.5991 |
2.0543 |
5-nonanone |
NONANONE |
10.6000 |
2.0150 |
n-octane |
OCTANE |
1.9406 |
1.9527 |
1-octanol |
OCTANOL |
9.8629 |
2.0435 |
2-octanone |
OCTANON2 |
9.4678 |
2.0025 |
n-pentadecane |
PENTDECN |
2.0333 |
2.0492 |
pentanal |
PENTANAL |
10.0000 |
1.9444 |
n-pentane |
NPENTANE |
1.8371 |
1.8428 |
pentanoic acid |
PENTACID |
2.6924 |
1.9839 |
1-pentanol |
PENTANOL |
15.1300 |
1.9884 |
2-pentanone |
PENTNON2 |
15.2000 |
1.9307 |
3-pentanone |
PENTNON3 |
16.7800 |
1.9388 |
1-pentene |
PENTENE |
1.9905 |
1.8810 |
E-2-pentene |
E2PENTEN |
2.0510 |
1.9025 |
pentyl acetate |
PENTACET |
4.7297 |
1.9664 |
pentylamine |
PENTAMIN |
4.2010 |
2.0967 |
perfluorobenzene |
PFB |
2.0290 |
1.8981 |
phenylmethanol |
BENZALCL |
12.4570 |
2.3704 |
propanal |
PROPANAL |
18.5000 |
1.8594 |
propanoic acid |
PROPACID |
3.4400 |
1.9235 |
1-propanol |
PROPANOL |
20.5240 |
1.9182 |
2-propanol |
PROPNOL2 |
19.2640 |
1.8978 |
propanonitrile |
PROPNTRL |
29.3240 |
1.8646 |
2-propen-1-ol |
PROPENOL |
19.0110 |
1.9980 |
propyl acetate |
PROPACET |
5.5205 |
1.9160 |
propylamine |
PROPAMIN |
4.9912 |
1.9238 |
pyridine |
PYRIDINE |
12.9780 |
2.2786 |
tetrachloroethene |
C2CL4 |
2.2680 |
2.2659 |
tetrahydrofuran |
THF |
7.4257 |
1.9740 |
tetrahydrothiophene-s,s-dioxide |
SULFOLAN |
43.9620 |
2.2002 |
tetralin |
TETRALIN |
2.7710 |
2.3756 |
thiophene |
THIOPHEN |
2.7270 |
2.3375 |
thiophenol |
PHSH |
4.2728 |
2.5259 |
toluene |
TOLUENE |
2.3741 |
2.2383 |
tributyl phosphate |
TBP |
8.1781 |
2.0232 |
1,1,1-trichloroethane |
TCA111 |
7.0826 |
2.0676 |
1,1,2-trichloroethane |
TCA112 |
7.1937 |
2.1650 |
trichloroethene |
TCE |
3.4220 |
2.1824 |
triethylamine |
ET3N |
2.3832 |
1.9628 |
2,2,2-trifluoroethanol |
TFE222 |
26.7260 |
1.6659 |
1,2,4-trimethylbenzene |
TMBEN124 |
2.3653 |
2.2644 |
2,2,4-trimethylpentane |
ISOCTANE |
1.9358 |
1.9363 |
n-undecane |
UNDECANE |
1.9910 |
2.0730 |
m-xylene |
M-XYLENE |
2.3478 |
2.2416 |
o-xylene |
O-XYLENE |
2.5454 |
2.2665 |
p-xylene |
P-XYLENE |
2.2705 |
2.2374 |
xylene (mixture) |
XYLENEMX |
2.3879 |
2.2485 |
1,1-dichloroethane |
10.1900 |
2.0880 |
|
1-iodopentene |
5.7800 |
2.2350 |
|
1-pentyne |
2.0600 |
1.9182 |
|
2-chlorobutane |
8.3900 |
1.9656 |
|
benzyl alcohol |
11.9200 |
2.3716 |
|
bromooctane |
5.0200 |
2.1083 |
|
butyl ethanoate |
5.0700 |
1.9432 |
|
butyl benzene |
2.3600 |
2.2201 |
|
carbon tet |
2.2300 |
2.1316 |
|
chlorotoluene |
6.8500 |
2.3654 |
|
decalin |
2.1900 |
2.1934 |
|
dimethylacetamide |
DMAC |
37.7800 |
2.0678 |
dimethylformamide |
DMF |
37.2200 |
2.0478 |
dimethylpyridine |
7.1700 |
2.2350 |
|
dodecane |
2.0100 |
2.0221 |
|
E-1,2-dichloroethene |
2.1400 |
2.0880 |
|
ethyl ethanoate |
6.0800 |
1.8824 |
|
ethyl methanoate |
8.3300 |
1.8469 |
|
ethyl eneglycol |
40.2500 |
2.0506 |
|
hexadecyl iodide |
3.5300 |
2.1934 |
|
hexanoic |
2.6000 |
2.0051 |
|
isobutanol |
16.7800 |
1.9460 |
|
isopropyl ether |
3.8800 |
1.8714 |
|
isopropyl toluene |
2.2300 |
2.2231 |
|
methyl ethanoate |
6.8600 |
1.8523 |
|
methyl methanoate |
8.8400 |
1.8036 |
|
methyl phenyl ketone |
17.4400 |
2.3624 |
|
methylformamide |
181.5600 |
2.0506 |
|
hexadecane |
2.0600 |
2.0592 |
|
methylaniline |
5.9600 |
2.4649 |
|
pentane |
1.8400 |
1.8414 |
|
pentadecane |
2.0300 |
2.0478 |
|
pentyl ethanoate |
4.7300 |
1.9656 |
|
phenyl ether |
3.7300 |
2.4932 |
|
propyl ethanoate |
5.5200 |
1.9155 |
|
pyrrolidine |
8.0400 |
2.0822 |
|
sec-butanol |
15.9400 |
1.9544 |
|
t-butanol |
12.4700 |
1.9238 |
|
t-butylbenzene |
2.3400 |
2.2290 |
|
tetrahyrothiophenedioxide |
43.9600 |
2.1993 |
|
tribromomethane |
4.2500 |
2.5632 |
|
trichloromethane |
TCM |
4.7100 |
2.0909 |
Z-1,2-dichloroethene |
9.2000 |
2.0996 |
|
isoquinoline |
11.0000 |
1.0100 |
|
quinoline |
9.1600 |
1.0100 |
|
diethylether |
4.2400 |
1.8295 |
|
dichloroethane |
10.1250 |
2.0874 |
|
carbontetrachloride |
2.2280 |
2.1319 |
|
heptane |
1.9113 |
1.9260 |
|
dimethylsulfoxide |
46.8260 |
2.0079 |
|
argon |
1.4300 |
1.4300 |
|
krypton |
1.5190 |
1.5190 |
|
xenon |
1.7060 |
1.7060 |
|
n-octanol |
9.8629 |
2.0435 |
|
aceticacid |
6.2528 |
1.8824 |
|
a-chlorotoluene |
6.7175 |
2.3688 |
|
benzylalcohol |
12.4570 |
2.3704 |
|
butanoicacid |
2.9931 |
1.9544 |
|
butylethanoate |
4.9941 |
1.9435 |
|
carbondisulfide |
2.6105 |
2.6631 |
|
decalin-mixture |
2.1960 |
2.1106 |
|
dibutylether |
3.0473 |
1.9578 |
|
diethylsulfide |
5.7230 |
2.0822 |
|
diisopropylether |
3.3800 |
1.8712 |
|
dimethyldisulfide |
9.6000 |
2.3375 |
|
diphenylether |
3.7300 |
2.4923 |
|
E-1,2-dichloroethene |
2.1400 |
2.0892 |
|
E-2-pentene |
2.0510 |
1.9025 |
|
ethylethanoate |
5.9867 |
1.8832 |
|
ethylmethanoate |
8.3310 |
1.8493 |
|
ethylphenylether |
4.1797 |
2.2729 |
|
formicacid |
51.1000 |
1.8807 |
|
hexanoicacid |
2.6000 |
2.0059 |
|
methylbenzoate |
6.7367 |
2.2995 |
|
methylbutanoate |
5.5607 |
1.9260 |
|
methylethanoate |
6.8615 |
1.8534 |
|
methylmethanoate |
8.8377 |
1.8045 |
|
methylpropanoate |
6.0777 |
1.8975 |
|
n-methylaniline |
5.9600 |
2.4599 |
|
n-methylformamide-mixture |
181.5600 |
2.0503 |
|
n,n-dimethylacetamide |
37.7810 |
2.0678 |
|
n,n-dimethylformamide |
37.2190 |
2.0463 |
|
pentanoicacid |
2.6924 |
1.9839 |
|
pentylethanoate |
4.7297 |
1.9664 |
|
propanoicacid |
3.4400 |
1.9235 |
|
propylethanoate |
5.5205 |
1.9160 |
|
tetrahydrothiophene-s,s-dioxide |
43.9620 |
2.2002 |
|
tributylphosphate |
8.1781 |
2.0232 |
|
xylene-mixture |
2.3879 |
2.2485 |
|
Z-1,2-dichloroethene |
9.2000 |
2.0996 |